




2. 400037 重庆,陆军军医大学(第三军医大学)第二附属医院妇产科
2. Department of Gynecology and Obstetrics, Second Affiliated Hospital, Army Medical University (Third Military Medical University), Chongqing, 400037, China
遗传性痉挛性截瘫(hereditary spastic paraplegia,HSP)是一组累及皮质脊髓束的运动神经元疾病,表现为下肢明显的痉挛和无力,可于新生儿、儿童或成年期发病,为患者及其家庭以及公共卫生造成极大负担。该类疾病具有临床/遗传异质性,基因检测是诊断的金标准。然而不典型的临床症状、庞大的致病基因谱及致病基因分子量巨大,导致临床对该类疾病的诊治具有局限性。
研究结果表明,轴突功能障碍是该疾病的基础,主要表现为下肢进行性无力和肌萎缩等。SPG 11基因编码spatacsin蛋白,是一种在神经元轴突生长和细胞内货物运输中发挥作用的蛋白质。SPG 11基因突变(无意突变,插入或缺失,均表明一种功能丧失的机制)均表现为纯合或复合杂合状态,遵循常染色体隐性遗传方式,在小鼠神经元中敲除SPG 11会导致神经突的突触囊泡的减少和轴突顺行运输的中断。SPG 11基因突变患者源性神经元表现出类似的顺行轴突运输缺陷,以及神经突内大量包涵体、膜包围结构和液泡,提示膜细胞器运输的改变。该类病例发病率低,临床较少见。现报道1例SPG 11纯合缺失导致遗传性痉挛性截瘫。
本研究收集了1例临床疑诊为遗传性痉挛性截瘫的家系,通过全基因组测序和Sanger测序对患者进行了基因诊断,明确突变位点,同时,验证家系成员突变携带情况,以探讨该家系的临床特点、分子遗传学特征及基因诊断的临床意义。为家系中的成员提供基因诊断和遗传咨询,为研究遗传性痉挛性截瘫的发病机制和治疗提供理论依据。
1 材料与方法 1.1 家系调查患者家系来自西南地区,汉族,该家系共有5代成员,其中Ⅳ-3患有与患者相同疾病,且已去世;患者双亲为近亲婚配。先证者男性,29岁,Ⅳ-6,因语言迟缓、不能行走于2021年4月30日于陆军军医大学第二附属医院遗传咨询门诊就诊。通过详细询问家族史,绘制该家系系谱图(图 1)。

|
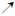 :示先证者
图 1
临床表现疑似遗传性痉挛性截瘫患者家系图 :示先证者
图 1
临床表现疑似遗传性痉挛性截瘫患者家系图
|
1.2 临床检查
在陆军军医大学第二附属医院遗传咨询门诊对该患者进行详细的临床病史和家族史调查,收集患者临床资料,包括体格检查、实验室检查、彩超、肌电图及颅脑MRI。
1.3 基因诊断本研究获得陆军军医大学伦理委员会批准(批件号:AMUWEC2022-10-01)。在取得患者及家系成员同意并签署知情同意书后,抽取先证者及其他10名家系成员外周抗凝静脉血2 mL,使用QIAmp DNA Blood Mini kit(Qiagen)试剂盒提取外周血基因组DNA。
1.3.1 二代测序通过超声剪切基因组DNA样品,然后将剪切的基因组DNA与Roche的NimbleGen 2.0捕获探针序列杂交。首先使用Agilent Bioanalyzer 2100测试文库的qPCR富集、大小分布和浓度,然后将样品在Illumina Hiseq2500上测序,对每个样品进行两次平行反应。按照制造商的说明书(Illumina),通过Illumina hiseq2500平台对富集的DNA进行测序。原始图像文件由BclToFastq(Illumina)处理以进行碱基调用并生成原始数据,使用质量得分≥20(Q20)过滤掉低质量的变异。使用BWA将测序读数与NCBI人参考基因组(hg19)比对。分别运用Samtools和Pindel分析序列的SNP和插入缺失。去除轻微等位基因频率(MAF)高于5%的同义变化和单核苷酸多态性(SNPs)(http://www.ncbi.nlm.nih.gov/projects/SNP);使用SIFT软件过滤掉非同义变化(http://sift.jcvi.org);然后分析突变基因的功能及其与疾病的关系。
1.3.2 一代测序(Sanger法)验证突变使用Sanger测序验证患者及家系成员突变携带情况,PCR引物(由擎科科技公司合成,产物长度为534 bp):SPG 11 -F,ATAATGGGGGCTTGCCTCAG(退火温度为60 ℃);SPG 11 -R,AAAGGTAGCCAGTGGTCAGT(退火温度为50 ℃)。50 μL反应体系行PCR扩增(北京天根),产物用2%琼脂糖凝胶电泳进行检测,ABI3130遗传分析仪直接测序。
2 结果 2.1 患者临床表现患者男性,29岁,表现为进行性下肢痉挛与无力,目前已丧失行走能力。患者幼时发育稍差、语言迟缓、四肢活动笨拙、控制较差、行走时双膝内翻跛行,随着年龄增长其症状逐渐加重,同时伴有双膝外侧肌肉僵硬。患者25岁时已无法完整表达,且行动笨拙,双下肢僵硬,无力,完全丧失行走能力。神经系统查体结果:患者智力正常、神志清楚、语言迟缓,躯干无肌萎缩,双下肢肌张力升高,双下肢近端肌力2级、远端肌力1级,行走不能。共济检查不配合,双侧深浅感觉正常。双上肢腱反射正常,双下肢腱反射亢进(++++)。双侧病理征未引出,脑膜刺激征阴性。患者父母为近亲婚配(图 1),家系中姐姐(Ⅳ-3)有相似临床表现,但未明确诊断,已去世。
2.2 辅助检查结果 2.2.1 实验室检查血常规、贫血三项、甲功、HIV、梅毒、同型半胱氨酸: 肌酸激酶、肌酸激酶同工酶、乳酸脱氢酶等未见异常。
2.2.2 肌电图下肢神经源性损害。
2.2.3 影像学心脏彩超、腹部彩超、双肾及双侧肾上腺区未见明显异常。颈椎MRI检查未见明显异常。颅脑MRI提示:小脑发育不良,胼胝体薄(图 2)

|
| 图 2 患者颅脑MRI部分影像结果 |
2.3 致病基因检测
患者全基因组测序结果显示,患者携带SPG 11基因致病性双等位缺失c.733_734del(p.M245Vfs*2),家系成员中,7名(Ⅰ-2、Ⅱ-2、Ⅱ-3、Ⅲ-1、Ⅲ-2、Ⅳ-5、Ⅴ-3) 携带此变异(图 3、4),患者父母均为携带者,为近亲婚配。

|
| 图 3 基因检测后明确诊断遗传性痉挛性截瘫11型患者家系图谱 |

|
| A:家系中正常对照测序图(Ⅳ-1);B:先证者测序图(Ⅳ-6);C:家系中突变携带者测序图(Ⅳ-5,Ⅴ-3) 图 4 患者及家系成员SPG 11基因Sanger测序 |
2.4 突变有害性分析
本例患者SPG 11基因突变为c.733_734delAT (exon4,NM_025137),导致氨基酸改变p.M245Vfs*2,为移码突变,产生了提前终止密码子。该突变位点在物种中高度保守,该变异在GnomAD种群数据库中频率较低,为0.000 3%,根据ACMG指南,该变异判定为致病性变异(Pathogenic)PVS1+PM3_Strong+PM2:PVS1:该变异为零效变异(移码突变),可能导致基因功能丧失;PM3_Strong:文献数据库已有该位点(Spastic paraplegia,autosomal recessive)隐性遗传的病例报道,变异标签为DM(致病突变),PM2:在正常人群数据库中的频率为0.000 3,为低频变异。
2.5 鉴别诊断本例患者主要表现为双下肢肌无力及肌张力增高,无感觉异常,根据该患者疾病特点,主要需与以下疾病鉴别:①肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS):肌萎缩性侧索硬化症是一种影响大脑中的上位运动神经元以及脑干和脊髓中的下位运动神经元的神经退行性病症,会导致瘫痪,无感觉异常。该病早期症状轻微,渐渐进展为全身性肌肉萎缩和吞咽困难,最后产生呼吸衰竭。该病的病理标志包括由运动神经元损失导致的皮质脊髓束苍白、残存运动神经元内出现的阳性泛素包涵体和病理性聚合物的沉积。本例患者仅表现为双下肢肌肉进行性无力及萎缩,未涉及上肢及躯干肌肉,也无呼吸困难等症状,故不支持该诊断。②双侧锥体束病损:为上运动神经元损害出现的原始反射。主要是指大脑皮质中的椎体细胞及其轴突和脊髓联系的一种网络系统,支配四肢及躯干运动。当锥体束病损时,失去了对脑干和脊髓的抑制功能而释放出踝和踇趾背伸的反射作用。查体时可出现肌张力增高,可能呈折刀样强直;腱反射活跃或亢进;病理反射阳性:做病理反射时,如Babinski征、Pussep征、Chaddock症、Oppenheim征、Gordon等检查阳性,可能会发现患者大拇趾背屈,四肢呈扇形张开,以及缝匠肌收缩等典型病理反射。本例患者双下肢肌肉痉挛,腱反射亢进,但病理征均为引出,故不支持该诊断。
2.6 SPG 11基因已知突变与表型分析SPG 11基因的100多个致病突变已被确认,我们通过文献检索与资料收集,分析了64种突变与表型的关系(表 1)。在这64种突变中,有5个剪接突变,18个无义突变,39个插入或缺失,这些突变均产生提前出现的终止密码子,导致蛋白质的截断,从而失去功能而致病。这些突变的致病模式中纯合致病占47%,复合杂合致病占15%,杂合致病占38%(图 5)。
| 突变类型 | 核苷酸/氨基酸突变位置 | 纯合/杂合状态 |
| Nonsense | c.118C>T/p.Q40X | |
| c.2198T>G/p.L733X | ||
| c.2697G>A/p.W899X | ||
| c.4668T>A/p.Y1556X | ||
| c.5970C>G/p.Y1990X | 纯合 | |
| c.5977C>T/p.E1993X | ||
| c.6091C>T/p.R2031X | ||
| c.6100C>T/p.R2034X | ||
| c.7023C>A/p.Y2341X | ||
| c.268G>T/p.E90X | ||
| c.751C>T/p.Q251X | ||
| c.1679C>G/p.S560X | ||
| c.1951C>T/p.R651X | 杂合 | |
| c.4306C>T/p.Q1436X | ||
| c.5470C>T/p.R1824X | ||
| c.5870C>G/p.S1957X | ||
| c.6856C>T/p.R2286X | ||
| c.6856C>T/p.R2286X, | ||
| c.2863delG/p.E955KfsX | 复合杂合 | |
| c.6856C>T/p.R2286X,c.2316+5G>A | ||
| Insertion/deletion | c.408_428del21/p.E136_I142del | |
| c.529_533del5/p.I177_F178del>S177fsX | ||
| c.2849_2850insT/p.L950FfsX | ||
| c.1697_1712del16insTACTCCCAT/p.D566VfsX | 杂合 | |
| c.1697_1712del16insTACTCCCAT/p.D566VfsX | ||
| c.4307_4308del2/p.Q1436RfsX | ||
| c.5992insT/p.Y1998fsX | ||
| c.3075_3076insA/p.E1026RfsX | ||
| c.5987_5990dupCTCT/p.C1996fsX | ||
| c.5255delT/p.F1752SfsX | ||
| c.3741_3742insA/p.P1248TfsX | ||
| c.5986_5987insT/p.C1996LfsX | ||
| c.733_734del2/p.M245VfsX, | ||
| c.5974C>T/p.R1992X | ||
| c.1203delA/p.K401fsX415, | ||
| c.2842_2843insG/p.V948GfsX | ||
| c.1551_1552delTT/p.C518SfsX, | 复合杂合 | |
| c.5867-1G>T (IVS30-1G > T)/p.Y1956RfsX | ||
| c. 453dupA/p.L152IfsX, | ||
| c.663G>A/p.W221X | ||
| c.6194C>G/p.S2056X, | ||
| c.5121+1C > T | ||
| c.5121_5122insAG/p.I1708RfsX, | ||
| c.6859C>T/p.Q2287X | ||
| c.733_734del2/p.M245VfsX | ||
| c.654_655delinsG/p.S218RfsX | ||
| c.1755_1758delAGCA/p. P585PfsX | ||
| c.1845_1848delGTCT/p.F617Lfs*5 | ||
| c.2163_2164insT/p.I722YfsX | ||
| c.7088_7089insATTA/p.Y2363X | ||
| c.6898_6899delCT/p.L2300AfsX | ||
| c.2355_2356del2/p.K785SfsX | ||
| c.2716delC/p.Q906SfsX | 纯合 | |
| c.2983_2984delTA/p.L934LfsX | ||
| c.3662_3665delTCAA/p.I1221RfsX | ||
| c.3719_3720delTA/p.I1240VfsX | ||
| c.4561delT/p.S1521EfsX | ||
| c.5898+5493_6509-491del/p.T1966fsX | ||
| c.6451delG/p,A2151PfsX | ||
| c.6790_6791insC/p.L2264PfsX | ||
| c.7029_7030insT/p.V2344CfsX | ||
| c.7101_7102insT/p.K2368X | ||
| Splice-site | c.2444G>T/p.R815M,r.? | |
| c.2444+1G>C,r.? | 杂合 | |
| c.6478+1G>T | ||
| c.2833A>G/p.R945GfsX, | 复合杂合 | |
| r.2834_2835ins2834+1_2834+65 | ||
| c. 4744-1G>A | 纯合 | |
| c.7151+4_7151+7delAGTA/p.K2384fsX | ||
| Missense | c.4046T>A/p.F1349I | 杂合 |
| c.833A>G/p.N278S | 纯合 |

|
| A:不同致病模式所占比例;B:不同突变类型所占比例 图 5 SPG 11基因已知突变类型统计 |
SPG 11基因突变导致的遗传性痉挛性截瘫11型为ARHSP最常见的类型,多呈复杂型表现,患者主要表现为儿童或青少年期发病,双下肢渐进性痉挛无力,伴认知受累,60%患者伴有构音障碍,共济失调等临床表现,90%患者颅脑磁共振显示薄型胼胝体。我们分析发现,15%以复合杂合模式致病的患者,临床表现更为严重,表现为发病年龄更早,病情进展快,最终均丧失站立及行走能力等[1-2]。
3 讨论人类SPG 11基因位于15q21.1染色体上,全长8 kb,包含40个外显子,编码spatacsin蛋白,主要有三种转录本,在中枢神经系统中均表达,空间转录组显示其全长转录本(~8 kb)主要表达于小脑,约5 kb的转录本主要表达于大脑皮层,spatacsin蛋白主要定位于神经元核周及膜上,在神经元轴突、树突以及突触连接处聚集[2]。SPG 11基因的100多个致病变异已在整个基因中被确定,绝大多数SPG 11突变都是移码突变或无义突变,这意味着SPG 11编码的蛋白质被截断,并且很可能失去功能。现有的体内和体外模型通过研究蛋白质在脂质清除过程中的作用将spatacsin蛋白的功能与发育和神经变性联系起来,脂质清除过程是由自噬-溶酶体机制介导。然而,关于蛋白质的确切定位和功能的许多问题仍不清楚,主要是由于蛋白质检测的难度大和对其结构的了解有限[3]。SPG 11 c.733_734delAT (p.Met245Valfs*2)突变导致提早终止密码子,预计将导致编码蛋白的截断或功能丧失介导的衰变而导致蛋白的缺失,这是疾病的常见机制。研究人员发现,p.Met245Valfs*2变异可纯合致病,也可与另一外显子上的错义突变c.5974C>T(p.R1992X)构成复合杂合模式致病,更有趣的是,当p.Met245Valfs*2纯合突变并带有一c.5974C>T(p.R1992X)杂合错义突变时,患者临床症状更显著[2],这种现象产生的原因仍有待于进一步研究(图 6)。开发可靠的可检测spatacsin蛋白表达的替代技术对于分析其时空表达、确定其功能和解释疾病表型至关重要。

|
| 图 6 SPG 11基因模式图 |
SPG 11的双等位致病变异是复杂HSP最可能的原因。SPG11-HSP患者以进行性痉挛和四肢麻痹为主要症状。其他症状根据中枢神经系统(central nervous system,CNS)或周围神经系统(peripheral nervous system,PNS)受累情况进行分类。CNS相关症状包括认知障碍、帕金森病、精神病和视觉障碍,而PNS相关症状包括神经病变和括约肌障碍;其他体征如外周淋巴水肿和肥胖也经常出现[3]。SPG 11突变也会导致早发常染色体隐性遗传肌萎缩性侧索硬化症(amyotrophic lateral sclerosis,ALS)和腓骨肌萎缩症(charcot-marie-tooth,CMT)。SPG11-HSP除了上运动神经元表型外,还经常表现为帕金森病,即“pallido-pyramidal syndrome(PPS)”;ALS是一种上、下运动神经元同时受累的神经系统变性疾病[4]。SPG11-ALS是一种罕见的疾病,发生在25岁之前。其特点是肢体和面部肌肉痉挛,伴有远端手足肌萎缩。它遵循一个缓慢的进展过程,患者的生存时间延长超过30年。CMT是一组成为遗传性感觉和运动神经病的病症,SPG11-CMT主要表现为单纯的下运动神经元表型,这些患者均未表现出痉挛、视网膜疾病或小脑功能障碍的迹象[5]。因此,SPG 11突变导致的遗传性痉挛性截瘫在诊断时需根据怀孕和分娩史,典型的症状和体征,疾病的进展速度,影像学检查,结合基因检测,与ALS和CMT进行鉴别诊断。
与单纯的HSP相比,复杂的SPG11-HSP开始于生命早期,常在10~20岁被诊断。其临床表现主要包括儿童时期开始的神经发育缺陷,最终会出现一系列广泛的症状,从简单的学习困难到严重的智力残疾。由于广泛的表型,许多患者在儿童早期没有被诊断出来,此时运动症状轻微或尚未出现。在结构层面上,胼胝体变薄是该疾病的主要表型标志[6],约90% 的SPG11-HSP患者影像学检查提示薄胼胝体。目前尚不清楚胼胝体薄的表型是先天性发育不良的结果,还是与渐进性萎缩有关,有关机制仍有待研究。然而,有研究认为,spatacsin蛋白的缺失改变了GSK3信号通路的平衡,使得GSK过度激活,而GSK的激活又促进了神经祖细胞(neural progenitor cells,NPCs)的增殖、神经发生和神经元的成熟,导致皮层NPCs自我更新能力受损;由于对称分裂的减少和不对称分裂的增加而导致皮层神经母细胞和神经元的过早生成,这些过程均依赖于受损的GSK3信号通路的调节,从而导致神经元存活率逐渐降低[7],这为薄胼胝体的机制研究提供了一个方向。迄今为止的实验支持了这样一种观点:由于投射神经元的逐渐退化,薄胼胝体的神经发育起源随着时间的推移而恶化。颅脑磁共振成像显示出胼胝体变薄,并结合病史,可以确定HSP的诊断。
研究人员在不同的SPG11-HSP模型中均观察到自噬相关的神经元缺陷。神经病理学研究显示,SPG11-HSP患者延髓和脊髓神经元中存在泛素和p62阳性颗粒,而p62蛋白的增加往往与自噬的减少相关[8],这可能是由于自噬溶酶体重组(autophagic lysosomal reformation,ALR)过程中的缺陷而导致的,而ALR对自噬过程中溶酶体内稳态的维持至关重要。研究发现spatacsin及其相互作用蛋白spastizin与ALR启动有关[9]。ALR损伤也能解释SPG11-HSP模型中自噬体累积和溶酶体增大的现象。溶酶体畸变和随后的脂质积累可能是由于spatacsin通过与spastizin和AP-5的相互作用引起的[10],AP-5与核内体到反高尔基体网络循环的甘露醇-6-磷酸受体(cation independent mannose-6-phosphate receptor,CIMPR)结合发挥作用,CIMPR是将溶酶体酶靶向到溶酶体的关键蛋白。事实上,阻断从核内体到高尔基体网络的运输可导致脂质积累,类似于与SPG11相关的神经退行性过程。自噬-溶酶体机制的功能障碍可能是SPG11突变蛋白在神经发育和神经退行性变中的一个致病机制。神经元是有丝分裂后的细胞,它们的正常功能高度依赖自噬过程,因此极易受到自噬过程异常的影响。有趣的是,在单纯遗传性痉挛性截瘫类型中也有溶酶体功能改变的报道,包括SPAST、ATL1和REEP1等基因突变导致的HSP[11]。溶酶体病理也涉及大多数PPS的相应机制。总的来说,神经元的损害是累积的,损害产生的有害后果随着疾病的进展而变得越来越明显。
目前HPS尚无特效治疗方法,主要为对症治疗,每天保证必要营养素的摄取以维持机体所需营养。临床遵医嘱可用解痉类药物进行治疗,如巴氯芬片、地西泮片等,能有效缓解遗传性痉挛性截瘫引起的肌肉僵硬和疼痛,也可以遵医嘱服用盐酸乙哌立松片,具有松弛肌肉、扩张血管、改善肌肉紧张、增加血流供应的作用。一项基于患者iPSC来源的神经前体细胞的研究发现,SPG 11突变患者神经前体细胞增殖减少是由GSK3β活性增加导致β-catenin信号受损介导的[7]。研究人员在一名SPG 11患者的iPSC衍生的神经元系和基因编辑细胞系上测试了GSK3β抑制剂tideglusib[12],挽救了神经病变,减少了细胞死亡,提示tideglusib(或类似分子)可以作为治疗SPG11-HSP的候选药物,为HSP治疗提供了重要依据,但其能否用于临床还有待于进一步研究。
婴幼儿期发病的SPG11-HSP是一种罕见的运动神经元病,其涉及神经发育障碍和神经退行性改变,由于其临床异质性,易造成疾病误诊。SPG 11基因双等位缺失突变c.733_734delAT (p.Met245Valfs*2)导致提前终止密码子,蛋白质发生截断而使细胞内蛋白质的缺失,使得神经细胞溶酶体功能异常及自噬改变,从而引起神经细胞发育异常及退行性改变,是目前认为导致遗传性痉挛性截瘫11型的主要原因。
本例患者提示我们,对有进行性双下肢萎缩伴无力、认知障碍,并伴有神经发育障碍的患者,应警惕此病的可能性。当影像学检查显示胼胝体变薄,应进行HSP相关基因Panel检测。此病目前尚无确切的治疗方法,因此,及早对HSP家系高危人群进行基因检测,明确患者后及早对症治疗,发现携带者后进行有效的产前诊断,对尽量延缓患者临床症状进程和预防HSP出生缺陷发生具有重要意义。
| [1] |
DENORA P S, SCHLESINGER D, CASALI C, et al. Screening of ARHSP-TCC patients expands the spectrum of SPG11 mutations and includes a large scale gene deletion[J]. Hum Mutat, 2009, 30(3): E500-E519. |
| [2] |
STEVANIN G, SANTORELLI F M, AZZEDINE H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum[J]. Nat Genet, 2007, 39(3): 366-372. |
| [3] |
POZNER T, REGENSBURGER M, ENGELHORN T, et al. Janus-faced spatacsin (SPG11): involvement in neuro-development and multisystem neurodegeneration[J]. Brain, 2020, 143(8): 2369-2379. |
| [4] |
KHANI M, SHAMSHIRI H, FATEHI F, et al. Description of combined ARHSP/JALS phenotype in some patients with SPG11 mutations[J]. Mol Genet Genomic Med, 2020, 8(7): e1240. |
| [5] |
MONTECCHIANI C, PEDACE L, LO GIUDICE T, et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease[J]. Brain, 2016, 139(Pt 1): 73-85. |
| [6] |
PASCUAL B, DE BOT S T, DANIELS M R, et al. Ears of the Lynx MRI sign is associated with SPG11 and SPG15 hereditary spastic paraplegia[J]. Am J Neuroradiol, 2019, 40(1): 199-203. |
| [7] |
MISHRA H K, PROTS I, HAVLICEK S, et al. GSK3ẞ-dependent dysregulation of neurodevelopment in SPG11-patient induced pluripotent stem cell model[J]. Ann Neurol, 2016, 79(5): 826-840. |
| [8] |
DENORA P S, SMETS K, ZOLFANELLI F, et al. Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions[J]. Brain, 2016, 139(Pt 6): 1723-1734. |
| [9] |
HIRST J, HESKETH G G, GINGRAS A C, et al. Rag GTPases and phosphatidylinositol 3-phosphate mediate recruitment of the AP-5/SPG11/SPG15 complex[J]. J Cell Biol, 2021, 220(2): e202002075. |
| [10] |
HIRST J, ITZHAK D N, ANTROBUS R, et al. Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval[J]. PLoS Biol, 2018, 16(1): e2004411. |
| [11] |
DENEUBOURG C, RAMM M, SMITH L J, et al. The spectrum of neurodevelopmental, neuromuscular and neurodegenerative disorders due to defective autophagy[J]. Autophagy, 2022, 18(3): 496-517. |
| [12] |
SHRIBMAN S, REID E, CROSBY A H, et al. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches[J]. Lancet Neurology, 2019, 18(12): 1136-1146. |