



2. 400038 重庆,陆军军医大学(第三军医大学)药学与检验医学系药物化学教研室
2. Department of Medicinal Chemistry, Faculty of Pharmacy & Laboratory Medicine, Army Medical University (Third Military Medical University), Chongqing, 400038, China
免疫检查点分子是机体监视肿瘤细胞体内生长增殖的重要屏障,由于机体免疫信号通路错综复杂,而且肿瘤微环境中免疫细胞、细胞因子、免疫佐剂等相互影响,因此仅对单一靶点的药物治疗效果还有待提高,而且也容易引起免疫不良反应。吲哚胺2, 3-双加氧酶1(indoleamine 2, 3-dioxygenase 1,IDO1)是人体色氨酸代谢的限速酶,也是调节肿瘤免疫应答的关键性免疫抑制酶,通过分解免疫细胞所需的关键氨基酸(色氨酸)抑制免疫细胞增殖,进而阻止人体对癌细胞发生的自然免疫反应[1-2]。IDO1通常在肿瘤细胞中过度表达,被认为是肿瘤免疫疗法的潜在药物靶点。IDO1抑制剂能够抑制色氨酸代谢提高免疫应答,从而达到抑制肿瘤的目的[3-4]。截至目前,尽管尚未有IDO1抑制剂上市,但已有多个抑制剂进入了临床研究阶段,包括Epacadostat[5-6]、Indoximod[7]、GDC-0919[8-9]和BMS-986205[10]等。这些抑制剂结构主要局限于吲哚类[11]、芳基咪唑类[12]及N-羟基脒类[13]等。为了进一步丰富IDO1抑制剂的结构类型,探究其构效关系,本研究根据IDO1抑制剂与IDO1蛋白晶体之间的结合模式及基于配体结构的设计,通过分子对接及药效团模型构建,对ZINC和Chembridge数据库进行虚拟筛选,结合体外酶活性验证以及分子动力学模拟,期望得到一种新型的小分子IDO1抑制剂。
1 材料与方法 1.1 材料 1.1.1 数据来源本研究所选取的人源IDO1蛋白晶体结构来源于PDB蛋白数据库(http://www.rcsb.org/pdb, ID: 4U72)。虚拟筛选的小分子来源于ZINC(http://zinc.docking.org/)、Chembridge(http://www.chembridge.com/)两个数据库。用于活性测试的化合物通过上海百灵威科技公司代购。
1.1.2 试剂及仪器磷酸氢二钾、磷酸二氢钾购自天津阿法埃莎公司;牛过氧化氢酶、抗坏血酸、亚甲基蓝三水化合物购自上海百灵威公司;IPTG,三氯乙酸购自美国Sigma公司;INCB024360购自美国BPS bioscience公司;大肠杆菌BL21(DE3)感受态细胞由陆军军医大学全军免疫学研究所提供;Ni-NTA亲和层析柱、Hiload 16/60 Superdex 200色谱层析柱购自美国GE Healthcare公司;酶标仪(SpectraMax® Paradigm® Multi-Mode Detection Platform)购自美国Molecular Devices公司;ChemiDocTM Touch Imaging System购自美国Bio-Rad公司。
1.2 方法 1.2.1 分子对接IDO1的蛋白晶体结构用SYBYL-X 2.0软件进行预处理:包括提取配体分子、删除水分子、加氢原子、去除氨基酸残基多重构象、加AMBER7 FF99力场、确定残基质子化状态,以及根据配体分子生成结合口袋等[14]。首先用“Lipinski”[15]对ZINC数据库的小分子进行初筛,并用MMFF94电荷和力场优化剩余小分子。使用Surflex-dock刚性对接程序对ZINC数据库小分子进行虚拟筛选,筛选对接得分大于8的化合物。随后对已筛选的小分子数据使用Surflex-dock进行柔性分子对接,根据配体分子与IDO1受体的对接得分、结合模式等综合确定先导化合物并开展生物学验证。
1.2.2 药效团模型的建立本研究收集文献报道已进入临床试验的3个IDO1小分子抑制,包括NLG-919[9]、Amg-1[16]和Epacadostat[6],结构如图 1所示。利用Discovery Studio 3.0软件的Pharmacophores模块构建药效团模型[17]。用MMFF94力场做能量优化,添加Principal全为2,最大可缺损性质(MaxOmitFeat)数目全为0,并利用Feature Mapping模块,选择氢键受体、氢键供体、疏水中心、正电荷中心、负电荷中心、芳环中心进行药效特征元素的提取,由BEST方法产生分子构象,基于Feature Mapping结果选择可能的药效团,最后通过HipHop算法进行特性结构比对,产生10个药效团模型。

|
| 图 1 进入临床研究的IDO1小分子抑制剂 |
为了保证药效团模型的可靠性,本研究从ChEMBL下载文献报道活性较好的80个IDO1小分子抑制剂,在Discovery Studio 3.0中对其加氢并进行结构优化。将已建立的10个药效团模型分别对80个小分子进行柔性匹配,使用Pharmacophores模块Ligand Profiler及Flexible/BEST方法,筛选匹配度高的药效团模型。基于药效团模型和IDO1小分子抑制剂的匹配热图,分析每个配体分子与不同药效团的匹配情况,确定最优药效团模型。
1.2.3 分子动力学建立先导化合物与IDO1对接得到的复合物进行分子动力学模拟[18]。将与抑制剂体系复合的IDO1受体置于0.15 mol/L NaCl溶液中,形成立方水箱,其中包括26 481个水分子,72个Na+离子和82个Cl-离子。蛋白质受体和配体的初始构型取自对接研究,初始模拟盒的尺寸为100 Å×100 Å×100 Å。
AMBER ff14SB力场[19]应用于蛋白质。水分子用TIP3P(可转移的中间电位,3点)水模型处理[20],配体的部分原子电荷使用半经验的键合电荷校正(AM1-BCC)方法得出[21-22]。从AFFER16中的GAFF获得其他力场参数[22]。使用ANTECHAMBER模块制备配体的残基拓扑结构。
使用AMBER16[23-24]包中的PMEMD.mpi和PMEMD.cuda模块进行MD模拟。首先,对系统进行能量最小化,以避免可能的空间碰撞。而后用2 fs的时间步长加热阶段,体系从0 K逐渐升温到300 K,2 fs及恒温(300 K)用于平衡阶段动力学模拟阶段。采用周期性边界条件来保持恒定的温度及压力,将压力设定为1atm并通过各向异性(x-,y-,z-)压力缩放法控制,压力弛豫时间为1ps。使用Langevin动力学调节温度,碰撞频率为2 ps-1[25-26]。采用粒子网格Ewald (PME)方法[27-28]处理远程静电,并设置10截止值来处理真实空间相互作用。所有涉及氢原子的共价键都利用SHAKE算法约束[29]。每个系统都经过100 ns的MD模拟,模拟系统的轨迹每100 ps保存1次。
1.2.4 重组IDO1酶的表达与纯化将人源IDO1全长编码序列插入到含硫氧还原蛋白(Trx)标签的表达载体pET32M3C中[30]构建pET32M3C-hIDO1重组质粒。经测序验证后转入大肠杆菌BL21(DE3)感受态细胞中,涂布于含50 μg/mL卡那霉素的LB琼脂平板上,挑取单菌落接种于含50 μg/mL卡那霉素的LB培养基中,37 ℃振荡培养至D(600)值在0.6~0.8之间,然后加入终浓度为0.5 mmol/L的5-氨基乙酰丙酸,30 ℃继续培养30 min,最后加入浓度为0.2 mmol/L的IPTG,16 ℃诱导表达18 h。将收集的菌体超声破碎后离心取得的上清液,经Ni离子亲和层析和Superdex 200凝胶过滤层析纯化后,得到重组人源IDO1蛋白。SDS-PAGE电泳鉴定目的蛋白表达情况,并用ChemiDocTM Touch Imaging System曝光显影,BCA法测定IDO1重组蛋白浓度。
1.2.5 IDO1酶活性测定本研究根据IDO1酶催化L-色氨酸产生犬尿氨酸的生物途径检测IDO1抑制剂活性(图 2),其原理如下:含铁血红素的IDO1与氧分子共同作用,催化L-Trp吲哚环的2, 3-双键氧化裂解,产生N-甲酰-L-犬尿氨酸,随后通过甲酰胺酶去甲酰化产生重要的代谢物L-犬尿氨酸,其与4-二甲氨基苯甲醛的乙酸溶液反应产生黄颜色反应,可用酶标仪在480 nm检测光密度值D(480)[31]。

|
| 图 2 IDO1催化L-色氨酸生成犬尿氨酸的生物途径 |
在100 μL的反应体系中,先将50 mmol/L磷酸钾缓冲溶液(pH=6.5),20 mmol/L抗坏血酸(用NaOH中和),10 μmol/L亚甲基蓝,200 μg/mL过氧化氢酶,50 nmol/L的IDO1酶,100 μmol/L L-色氨酸混合后,再加入1 nmol/L~100 μmol/L的待测化合物,37 ℃反应30 min。加入40 μL 30%的三氯乙酸终止反应,65 ℃孵育15 min,12 000 r/min离心10 min,取100 μL的上清液于96孔板中,加入100 μL的4-二甲氨基苯甲醛乙酸溶液(2%),室温摇床上孵育10 min,酶标仪于480 nm处检测光密度值。根据如下公式将测得的光密度值转换为抑制率:
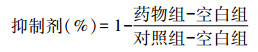
其IC50值用GraphPad Prism 7软件对浓度-抑制率四参数拟合得曲线。
2 结果 2.1 基于分子对接的虚拟筛选为了获得结构新颖的IDO1小分子先导化合物,本研究使用分子对接方法对ZINC小分子数据进行虚拟筛选。经过“Lipinski”规则和刚性分子对接筛选获得492个小分子,进一步的柔性对接确定11个候选小分子(表 1)。图 3A显示所筛选的小分子均可与IDO1受体蛋白在空间上很好的契合。其中,亲水区(空腔A)的亚铁血红素可以与配体螯合,对配体的活性具有决定性作用,位于疏水区(空腔B)对配体选择性具有重要作用。综合对接得分与结合模式发现,ZINC91657208与IDO1不仅可与亚铁血红素中的铁离子稳定络合(图 3B),而且可与残基Ser-167、Gly-236、Gly-262形成稳定的氢键。ZINC91657208与IDO1相互作用二维图(图 3C)显示,ZINC91657208苯并咪唑环上的羰基O作为氢键受体与Ser-167侧链形成氢键,喹啉环上的羰基O同样作为氢键供体与Gly-261、Phe-291主链形成氢键,喹啉环上的仲胺作为氢键供体与Gly-262主链形成氢键,ZINC91657208结构中的2个仲胺作为氢键受体均与亚铁血红素HEM-501形成氢键,结果表明进入A腔的4-羟基喹啉骨架是ZINC91657208的关键基团。
| ZINC ID | 结构 | 分子对接打分 | 氢键作用残基 | IC50±SD(μmol/L) |
| ZINC67909150 | 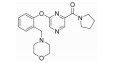
|
12.1869 | Ser-167 | >100 |
| ZINC91657208 | 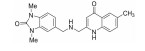
|
11.5085 | Ser-167、Gly-236、Gly-262 | 77.15±9.05 |
| ZINC67674024 | 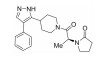
|
11.3822 | Gly-236 | >100 |
| ZINC72463748 | 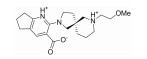
|
11.1228 | Gly-262、Ala-264 | >100 |
| ZINC63855889 | 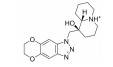
|
11.0281 | Ser-167、Gly-262 | >100 |
| ZINC95497050 | 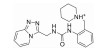
|
10.8507 | Gly-262 | >100 |
| ZINC95362825 | 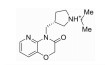
|
10.8014 | Ser-167、Gly-262 | >100 |
| ZINC96153859 | 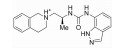
|
10.7367 | - | >100 |
| ZINC72168284 | 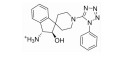
|
10.3513 | Arg-231、Ser-235 | >100 |
| ZINC65433995 | 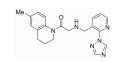
|
10.2781 | Ser-167、Ala-264 | >100 |
| ZINC72174209 | 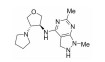
|
9.9541 | Gly-262 | >100 |

|
| A: 11个候选小分子与IDO1受体蛋白的对接叠合图,蓝色代表亲水性区域,黄褐色代表疏水性区域;IDO1晶体由亚铁红素结构和两个空腔(空腔A和空腔B)组成;其中,亲水区(空腔A)的亚铁血红素可以与配体螯合,对配体的活性具有决定性作用,位于疏水区(空腔B)对配体选择性具有重要作用;B: ZINC91657208与IDO1相互作用的三维图;C: ZINC91657208与IDO1蛋白相互作用的二维图,绿色圆圈表示范德华力作用,品红色圆圈表示氢键、电荷或极性相互作用,氨基酸外的蓝色光晕圈表示氨基酸相互作用的溶剂残留,直径大小与溶剂残留呈正比;黑色带箭头的虚线表示非氨基酸残基与配体相互作用的氢键,绿色带箭头的虚线表示氨基酸主链与配体相互作用的氢键,蓝色带箭头的虚线表示氨基酸侧链与配体相互作用形成的氢键,箭头指向电子供体 图 3 ZINC91657208与IDO1晶体的分子对接模型图 |
2.2 重组IDO1蛋白的表达和纯化
经IPTG诱导后SDS-PAGE分析结果显示(图 4),全菌蛋白在59×103出现明显的新增条带,与重组蛋白理论预测相对分子量相符。经镍柱亲和层析和分子筛层析纯化后,SDS-PAGE结果显示目的蛋白纯度较高。超滤离心浓缩后BCA法测定蛋白浓度为3.45 mg/mL。

|
| M:蛋白分子质量标准1:300 ng重组IDO1蛋白;2:3 μg重组IDO1蛋白;3:10 μg重组IDO1蛋白;4:未经IPTG诱导的pET32M3C-hIDO1超声破碎后沉淀;5:重组菌pET32M3C-hIDO1/BL21(DE3)经超声破碎后的上清 图 4 重组IDO1蛋白的SDS-PAGE电泳结果 |
2.3 IDO1抑制剂活性评价
通过IDO1活性检测体系,绘制酶反应时间曲线(图 5),验证了重组人源IDO1的活性。检测阳性化合物INCB024360的抑酶活性,IC50值为84.2 nmol/L,与文献[6]报道的67 nmol/L相近,表明该反应检测体系可靠有效。

|
| 图 5 IDO1酶反应时间曲线 |
使用该体系测定从ZINC数据库筛选获得小分子的IDO1抑制剂活性,结果显示ZINC91657208的抑制剂活性(77.15±9.05)μmol/L,其他分子的抑制活性均超过了100 μmol/L,结合模式显示其他分子与IDO1最多形成2个氢键,与ZINC91657208存在明显差别。为了寻找更优异的先导化合物,本研究基于ZINC91657208的药效团结构特征,进一步使用药效团模型对ChEMBL数据进行筛选。
2.4 药效团模型的构建及筛选本研究基于已进入临床研究的3个小分子化合物所建药效团模型,通过Feature Mapping共提取获得氢键受体(36)、氢键供体(17)、疏水中心(8)、正电荷中心(2)和芳环中心(12)共75个药效特征元素,结果如图 6所示。基于特征元素,利用3个IDO1抑制剂的结构比对构建10个药效团模型。为了获得优异的药效团模型用于虚拟筛选,将所建模型对ChEMBL的80个IDO1抑制剂小分子进行柔性匹配,匹配热图(图 7)显示第6个药效团模型与各个活性小分子匹配值最高,说明该药效团模型具有良好的预测能力。图 8显示该模型包括有1个芳香环特征,1个疏水特征以及3个氢键受体特征。Rank打分值为32.914,此模型特征与Amg-1和Epacadostat两个分子均可匹配,与NLG-919部分匹配。因此,本研究最终选择第6个药效团模型为预测模型。

|
| 绿色球表示氢键受体,紫色球表示氢键给体,蓝色球为疏水基团,红色为正电荷中心,黄色球为芳环中心 图 6 药效特征元素结果 |

|
| 横坐标表示10个药效团模型,纵坐标表示80个IDO1抑制剂小分子,分值的大小用不同颜色表示,从深红色为高,依次递减,蓝色为低 图 7 药效团模型和测试集小分子的匹配热图 |

|
| 灰色为NLG-919,紫色为Amg-1,黄色为Epacadostat 图 8 IDO1抑制剂最优药效团模型 |
基于分子对接从ZINC数据库筛选获得活性较好的小分子ZINC91657208,利用Chembridge数据库亚结构检索其活性部分4-羟基喹啉得到341个化合物,随后根据类药性五原则筛选出31个化合物。采用Search 3D Database模块,设置FAST构象,使用已验证的药效团模型6对31个化合物进行虚拟筛选。拟合值(Fit vable)评判数据库小分子与药效团模型匹配情况,最终筛选出拟合值高的10个化合物进行生物学验证(表 2)。进一步生物学活性评价显示,在100 μmol/L浓度下达到50%以上的抑制率的化合物有3个,其中Chembridge29374490的IC50约为37.78 μmol/L。
| Chembridge编号 | R1基团 | R2基团 | R3基团 | 抑制率(100 μmol/L) | IC50±SD(μmol/L) |
| 15974208 | 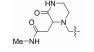
|
Cl | 25% | >100 | |
| 22054105 | 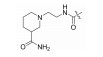
|
F | 65% | 88.96±15.25 | |
| 81785538 | 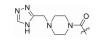
|
12% | >100 | ||
| 29374490 | 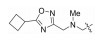
|
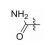
|
77% | 37.78±5.87 | |
| 52615985 | 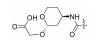
|
F | 13% | >100 | |
| 83771158 | 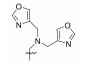
|
F | 33% | >100 | |
| 65969542 | 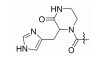
|
F | 16% | >100 | |
| 76920555 | 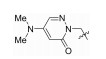
|
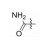
|
24% | >100 | |
| 78263001 | 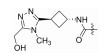
|
F | 5% | >100 | |
| 80455043 | 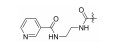
|
F | 18% | >100 | |
| 55846669Zinc91657208 | 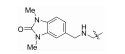
|
Me | 62% | 77.15±9.05 | |
4-羟基喹啉类IDO1抑制剂的结构及R1、R2、R3基团在其中的位置: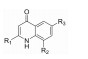
| |||||
2.5 分子动力学模拟
为进一步研究4-羟基喹啉类化合物与IDO1的动态相互作用,对先导化合物Chembridge29374490与IDO1进行了100 ns动力学模拟。在分子动力学模拟过程中,利用合适的力场、步长、模拟时间以及体系动力学过程的充分稳定平衡获得可靠的计算结果。
将Chembridge29374490与IDO1分子对接结果(绿色),即0 ns的动力学模拟和100 ns动力学模拟后(蓝色)的体系状态进行比对(图 9A),两者表现出良好的重叠性,且模拟后的Chembridge29374490结构显示与IDO1受体更近的结合距离。此外,对于Chembridge 29374490与IDO1体系的RMSD统计图(图 9B)显示,抑制剂的波动大于在1 (图中的红线),而受体始于3而后缓慢降至70 ns的2.3并在此范围波动,表明从0 ns到100 ns的模拟过程中保持相当稳定的状态。如图 9C所示,将Chembridge 29374490对接到IDO1的晶体结构中,结合口袋中涉及到的重要残基包括Try-126, Cys-129, Phe-163, Phe-164, Ser-167, Phe-226, Arg-231, Leu-234, Lys-238, Gly-262, Ser-263, Ala-264, Ile-354,这里描述的关键残基与从IDO1晶体结构获得的信息基本一致[32]。根据对接结果,可发现Ser-163通过氢键与Chembridge29374490相互作用。为进一步验证两者的相互作用,进行100 ns的动力学模拟,并生成最后20 ns的平均结构并用于分析(图 9D)。Chembridge 29374490在MD模拟期间相对稳定,具有明显的构象变化,例如,Lys-238与小分子形成氢键以增加结合稳定性。以上所有信息表明,Chembridge 29374490能够与IDO1稳定地相互作用。

|
| A:Chembridge29374490与IDO1动力学模拟前(分子对接结果,绿色)及100 ns动力学模拟后(蓝色)对比;B:Chembridge29374490-IDO1复合物的RMSD; C:Chembridge29374490与IDO1的相互作用(0 ns); D: Chembridge29374490与IDO1经100 ns动力学模拟后的相互作用 图 9 Chembridge29374490与IDO1的相互作用及分子动力学模拟 |
3 讨论
免疫检查点分子是介导肿瘤免疫逃逸的重要途径,是肿瘤免疫治疗的重要靶点[1-2]。本研究以免疫检查点分子IDO1为靶点,采用了以分子对接为主的筛选方法:首先使用刚性对接对ZINC的200万个化合物进行虚拟筛选,通过选取对接打分(Docking score)大于8的化合物,快速缩小筛选范围,再进一步使用柔性对接方法进行精确筛选。最后,通过手动筛选的方式,逐一考虑配体与受体构象的结构位置,仔细分析受体-配体分子间的相互作用,结合经验性判断及化合物的结构类型,筛选得到11个先导化合物。通过最后的生物活性验证,发现了新型的IDO1抑制剂ZINC91657208,其骨架结构类型为4-羟基喹啉类,而这一骨架结构类型同已报道的IDO1抑制剂不同,这将为后续的结构优化以发现新型的抗肿瘤药物奠定基础。同时,我们研究了ZINC91657208与IDO1受体的结合模型。IDO1受体的活性位点由两个结合口袋共同组成,其中一个与铁离子直接作用,另一个可以容纳较大空间体积的基团。通过分子对接研究,我们发现ZINC91657208喹啉环上的仲胺可以与亚铁血红素形成配位键,喹啉环上的羰基氧与Gly-261、Phe-291形成氢键作用,从而使4-羟基喹啉类化合物与IDO1蛋白的结合更加稳定。
为了进一步的优化先导化合物结合,本研究用已进入临床研究的3个小分子化合物,构建了IDO1抑制剂的药效团模型,其包括有1个芳香环特征,1个疏水特征以及3个氢键受体特征。根据该药效团模型,我们对化合物库进行再次筛选,选取了10个含有4-羟基喹啉类化合物骨架结构的类似物。经体外酶活性验证,筛选出3个能够明显抑制IDO1酶活性的化合物(Chembridge22054105,Chembridge29374490,Chembridge 55846669)。其中,Chembridge29374490的IC50大约为37.78 μmol/L,活性远高于文献报道的IDO1抑制剂1-MT(IC50=380 μmol/L)。最后,借助分子动力学模拟对高活性的Chembridge29374490与IDO1的相互作用模式进行研究。结果表明,该化合物能够与IDO1的Ser-163和Lys-238氨基酸残基形成稳定的氢键相互作用,使两者能够稳定结合。以上研究表明,4-羟基喹啉类化合物是一种新型的IDO1抑制剂。虽然该类化合物对IDO1的抑酶活性较低,但其结构新颖、易于进行后续的衍生,因此4-羟基喹啉类骨架IDO1抑制剂的发现,将为设计和发展抑制新型的IDO1抑制剂提供理论基础[3-4],有望获得具有知识产权的新型IDO1抑制剂及新型的肿瘤免疫治疗药物。
本研究将计算机辅助药物设计方法应用于药物先导化合物的快速发现,节省了先导化合物发现的时间,降低了筛选成本。经生物试验验证,确保了筛选结果的可靠性,达到发现新型IDO1抑制剂先导化合物的目的,并初步阐明了其构效关系及与IDO1晶体的结合位点,为后续的先导化合物结构的优化改造奠定了基础。
| [1] |
MUNN D H, MELLOR A L. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance[J]. Trends Immunol, 2016, 37(3): 193-207. DOI:10.1016/j.it.2016.01.002 |
| [2] |
LIU M, WANG X, WANG L, et al. Targeting the IDO1 pathway in cancer: from bench to bedside[J]. J Hematol Oncol, 2018, 11(1): 100. DOI:10.1186/s13045-018-0644-y |
| [3] |
PRENDERGAST G C, MALACHOWSKI W J, MONDAL A, et al. Indoleamine 2, 3-dioxygenase and its therapeutic inhibition in cancer[J]. Int Rev Cell Mol Biol, 2018, 336: 175-203. DOI:10.1016/bs.ircmb.2017.07.004 |
| [4] |
李湘, 樊钱永, 沈舜义. 吲哚胺2, 3-双加氧酶抑制剂及相关抗肿瘤药物开发[J]. 世界临床药物, 2017, 38(7): 483-488. LI X, FAN Q Y, SHEN S Y. Indoleamine-2, 3-dioxygenase inhibitor and its related anti-tumor drugs development[J]. World Clin Drugs, 2017, 38(7): 483-488. DOI:10.13683/j.wph.2017.07.012 |
| [5] |
LIU X D, SHIN N, KOBLISH H K, et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity[J]. Blood, 2010, 115(17): 3520-3530. DOI:10.1182/blood-2009-09-246124 |
| [6] |
BEATTY G L, O'DWYER P J, CLARK J, et al. First-in-human phase Ⅰ study of the oral inhibitor of indoleamine 2, 3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies[J]. Clin Cancer Res, 2017, 23(13): 3269-3276. DOI:10.1158/1078-0432.CCR-16-2272 |
| [7] |
SOLIMAN H H, MINTON S E, HAN H S, et al. A phase Ⅰ study of indoximod in patients with advanced malignancies[J]. Oncotarget, 2016, 7(16): 22928-22938. DOI:10.18632/oncotarget.8216 |
| [8] |
MAUTINO M R, JAIPURI F A, WALDO J, et al. Abstract 491: NLG919, a novel indoleamine-2, 3-dioxygenase (IDO)-pathway inhibitor drug candidate for cancer therapy[J]. Cancer Res, 2013, 73(8 Supplement): 491. DOI:10.1158/1538-7445.am2013-491 |
| [9] |
NAYAK-KAPOOR A, HAO Z L, SADEK R, et al. Phase Ia study of the indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors[J]. J Immunother Cancer, 2018, 6(1): 61. DOI:10.1186/s40425-018-0351-9 |
| [10] |
SIU L L, GELMON K, CHU Q, et al. Abstract CT116: BMS-986205, an optimized indoleamine 2, 3-dioxygenase 1 (IDO1) inhibitor, is well tolerated with potent pharmacodynamic (PD) activity, alone and in combination with nivolumab (nivo) in advanced cancers in a phase 1/2a trial[J]. Cancer Res, 2017, 77(13 Supplement): CT116. DOI:10.1158/1538-7445.am2017-ct116 |
| [11] |
QIAN S, HE T, WANG W, et al. Discovery and preliminary structure-activity relationship of 1H-indazoles with promising indoleamine-2, 3-dioxygenase 1 (IDO1) inhibition properties[J]. Bioorg Med Chem, 2016, 24(23): 6194-6205. DOI:10.1016/j.bmc.2016.10.003 |
| [12] |
KUMAR S, JALLER D, PATEL B, et al. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2, 3-dioxygenase[J]. J Med Chem, 2008, 51(16): 4968-4977. DOI:10.1021/jm800512z |
| [13] |
YUE E W, DOUTY B, WAYLAND B, et al. Discovery of potent competitive inhibitors of indoleamine 2, 3-dioxygenase with in vivo pharmacodynamic activity and efficacy in a mouse melanoma model[J]. J Med Chem, 2009, 52(23): 7364-7367. DOI:10.1021/jm900518f |
| [14] |
唐光辉, 张娅, 张玉萍, 等. 含磷嘧啶类CDK9抑制剂的分子对接、3D-QSAR和分子动力学模拟[J]. 高等学校化学学报, 2017, 38(11): 2061-2069. TANG G H, ZHANG Y, ZHANG Y P, et al. Molecular docking, QSAR and molecular dynamics simulation on phosphorus containing pyrimidines as CDK9 inhibitors[J]. Chem J Chin Univ, 2017, 38(11): 2061-2069. DOI:10.7503/cjcu20170237 |
| [15] |
张洁, 谭初兵, 徐为人, 等. Lipinski五规则的研究进展[J]. 药物评价研究, 2011, 34(6): 451-455. ZHANG J, TAN C B, XU W R, et al. Advances in studies on Lipinski's five rules[J]. Drug Eval Res, 2011, 34(6): 451-455. |
| [16] |
MEININGER D, ZALAMEDA L, LIU Y, et al. Purification and kinetic characterization of human indoleamine 2, 3-dioxygenases 1 and 2 (IDO1 and IDO2) and discovery of selective IDO1 inhibitors[J]. Biochim Biophys Acta, 2011, 1814(12): 1947-1954. DOI:10.1016/j.bbapap.2011.07.023 |
| [17] |
汪滢, 唐国荣, 刘澍楠, 等. 结合分子相似性、药效团和分子对接筛选新的HIV-1蛋白酶抑制剂[J]. 生物信息学, 2015, 13(4): 244-250. WANG Y, TANG G R, LIU S N, et al. Discovery of new HIV-1 protease inhibitors by integrating molecular similarity, pharmacophore and docking methods[J]. China J Bioinformatics, 2015, 13(4): 244-250. DOI:10.3969/j.issn.1672-5565.2015.04.07 |
| [18] |
李博, 周锐, 何谷, 等. 螺环吲哚类MDM2抑制剂的分子对接、定量构效关系和分子动力学模拟[J]. 化学学报, 2013, 71(10): 1396-1403. LI B, ZHOU R, HE G, et al. Molecular docking, QSAR and molecular dynamics simulation on spiro-oxindoles as MDM2 inhibitors[J]. Acta Chimica Sinica, 2013, 71(10): 1396-1403. DOI:10.6023/A13040375 |
| [19] |
MAIER J A, MARTINEZ C, KASAVAJHALA K, et al. Ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB[J]. J Chem Theory Comput, 2015, 11(8): 3696-3713. DOI:10.1021/acs.jctc.5b00255 |
| [20] |
JORGENSEN W L, CHANDRASEKHAR J, MADURA J D, et al. Comparison of simple potential functions for simulating liquid water[J]. J Chem Phys, 1983, 79(2): 926-935. DOI:10.1063/1.445869 |
| [21] |
JAKALIAN A, JACK D B, BAYLY C I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: Ⅱ. Parameterization and validation[J]. J Comput Chem, 2002, 23(16): 1623-1641. DOI:10.1002/jcc.10128 |
| [22] |
WANG J M, WOLF R M, CALDWELL J W, et al. Development and testing of a general amber force field[J]. J Comput Chem, 2004, 25(9): 1157-1174. DOI:10.1002/jcc.20035 |
| [23] |
GÖTZ A W, WILLIAMSON M J, XU D, et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born[J]. J Chem Theory Comput, 2012, 8(5): 1542-1555. DOI:10.1021/ct200909j |
| [24] |
SALOMON-FERRER R, GÖTZ A W, POOLE D, et al. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle mesh ewald[J]. J Chem Theory Comput, 2013, 9(9): 3878-3888. DOI:10.1021/ct400314y |
| [25] |
LONCHARICH R J, BROOKS B R, PASTOR R W. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide[J]. Biopolymers, 1992, 32(5): 523-535. DOI:10.1002/bip.360320508 |
| [26] |
IZAGUIRRE J A, CATARELLO D P, WOZNIAK J M, et al. Langevin stabilization of molecular dynamics[J]. J Chem Phys, 2001, 114(5): 2090-2098. DOI:10.1063/1.1332996 |
| [27] |
DARDEN T, YORK D, PEDERSEN L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems[J]. J Chem Phys, 1993, 98(12): 10089-10092. DOI:10.1063/1.464397 |
| [28] |
ESSMANN U, PERERA L, BERKOWITZ M L, et al. A smooth particle mesh Ewald method[J]. J Chem Phys, 1995, 103(19): 8577-8593. DOI:10.1063/1.470117 |
| [29] |
RYCKAERT J P, CICCOTTI G, BERENDSEN H J C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes[J]. J Compt Phys, 1977, 23(3): 327-341. DOI:10.1016/0021-9991(77)90098-5 |
| [30] |
CROSIGNANI S, BINGHAM P, BOTTEMANNE P, et al. Discovery of a novel and selective indoleamine 2, 3-dioxygenase (IDO-1) inhibitor 3-(5-fluoro-1H-indol-3-yl)pyrrolidine-2, 5-dione (EOS200271/PF-06840003) and its characterization as a potential clinical candidate[J]. J Med Chem, 2017, 60(23): 9617-9629. DOI:10.1021/acs.jmedchem.7b00974 |
| [31] |
BASRAN J, BOOTH E S, LEE M, et al. Analysis of reaction intermediates in tryptophan 2, 3-dioxygenase: A comparison with indoleamine 2, 3-dioxygenase[J]. Biochemistry, 2016, 55(49): 6743-6750. DOI:10.1021/acs.biochem.6b01005 |
| [32] |
CHENG M F, HUNG M S, SONG J S, et al. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2, 3-dioxygenase inhibitors[J]. Bioorg Med Chem Lett, 2014, 24(15): 3403-3406. DOI:10.1016/j.bmcl.2014.05.084 |